My major research interest is to understand biological processes using computational methods (QM calculations and molecular dynamics simulations). In particular, I study the following...
Brief description of works published till date:
Simulation of Pluronics in non-aqueous solvents.
Although there are a handful of models available for Pluronics, none of them are tested for their performance in non-aqueous solvents. As we intend to study the interaction of Pluronics with complex biological environments, it is important that we make sure that the model reproduces the properties of Pluronics in different environments. In this part of our study, we simulated the Pluronics and its components i.e., polyethylene oxide (PEO) and polypropylene oxide (PPO) in water, methanol, chloroform, carbon tetrachloride and n-heptane. The workflow started with studying the properties of the monomers of PEO and PPO i.e., dimethoxyethane (DME) and dimethoxypropane (DMP) in non-aqueous solvents. The the model satisfactorily reproduced the experimental data and helped us to find insights on the properties of the polymers.
- the translocation of antibiotics through bacterial porins,
- interaction of polymers with biological membranes,
- polymers as drug carriers,
- behaviour of polymers in solutions.
Brief description of works published till date:
Simulation of Pluronics in non-aqueous solvents.
Although there are a handful of models available for Pluronics, none of them are tested for their performance in non-aqueous solvents. As we intend to study the interaction of Pluronics with complex biological environments, it is important that we make sure that the model reproduces the properties of Pluronics in different environments. In this part of our study, we simulated the Pluronics and its components i.e., polyethylene oxide (PEO) and polypropylene oxide (PPO) in water, methanol, chloroform, carbon tetrachloride and n-heptane. The workflow started with studying the properties of the monomers of PEO and PPO i.e., dimethoxyethane (DME) and dimethoxypropane (DMP) in non-aqueous solvents. The the model satisfactorily reproduced the experimental data and helped us to find insights on the properties of the polymers.
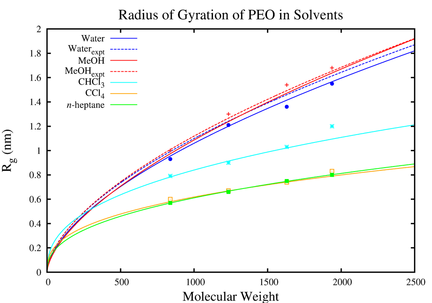

Fig: (Top )Radius of gyration for PEO18-43 plotted against molecular weight for all solvents at 298 K. (Bottom) Spatial density distributions of the solvent atoms in the first solvation shell of DME (upper row) and DMP (middle row) at 318 K. From the left to right, water (SPC), methanol, CCl4, and n-heptane density distribution are reported, respectively. The hydrogen atoms are in blue, oxygen in red, carbon in green, and chlorine in yellow. The density surfaces have isovalues of 0.012, 0.004, 0.0016, and 0.0008 for water, methanol, carbon tetrachloride, andn-heptane, respectively. In the lower row, spatial density distributions of the solvent atoms in the first solvation shell of DME in R-DMP (left) in racemic DMP (middle) and DMP in DME (right) at 318 K are shown. The oxygen atoms are in red and carbon in green. The density surfaces have isovalues of 0.005.
Diffusion of DME and DMP through DMPC lipid bilayers.
In this part of work, we theoretically studied DME and DMP at water/n-heptane and 1,2-dimyristoyl-sn-glycero-3-phospatidycholine (DMPC) lipid bilayer/water interfaces using the umbrella sampling method. The percolation free energy barrier of one DME and DMP molecule from water to n-heptane phase calculated using the umbrella sampling method turned out to be equal to 18.5 kJ/mol and 6 kJ/mol, respectively. In the case of the DMPC lipid bilayer, overall free energy barriers of 20 kJ/mol and 12 kJ/mol were obtained for DME and DMP, respectively. The spontaneous diffusion of DME and DMP in the lipid bilayer has also been investigated using unconstrained molecular dynamics simulations at the water/DMPC interface and inside the lipid bilayer. As expected from the estimated percolation barriers, simulation results show that DME, contrary to DMP, spontaneously diffuse into the aqueous solution from the lipid interior. In addition, simulations with multiple DME or DMP molecules at the interface show spontaneous diffusion within 50 ns inside the DMPC layer only for DMP.
In this part of work, we theoretically studied DME and DMP at water/n-heptane and 1,2-dimyristoyl-sn-glycero-3-phospatidycholine (DMPC) lipid bilayer/water interfaces using the umbrella sampling method. The percolation free energy barrier of one DME and DMP molecule from water to n-heptane phase calculated using the umbrella sampling method turned out to be equal to 18.5 kJ/mol and 6 kJ/mol, respectively. In the case of the DMPC lipid bilayer, overall free energy barriers of 20 kJ/mol and 12 kJ/mol were obtained for DME and DMP, respectively. The spontaneous diffusion of DME and DMP in the lipid bilayer has also been investigated using unconstrained molecular dynamics simulations at the water/DMPC interface and inside the lipid bilayer. As expected from the estimated percolation barriers, simulation results show that DME, contrary to DMP, spontaneously diffuse into the aqueous solution from the lipid interior. In addition, simulations with multiple DME or DMP molecules at the interface show spontaneous diffusion within 50 ns inside the DMPC layer only for DMP.
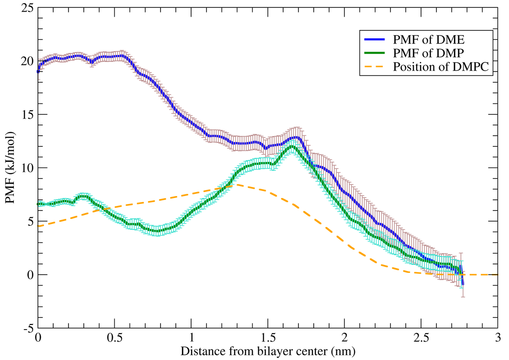
Fig: The comparison of PMF profiles of percolation of DME and DMP molecules using US method. The position of the DMPC bilayer is shown in orange.
Interaction of Curcumin with Pluronics.
Curcumin, a naturally occurring drug molecule, has been extensively investigated for its various potential usages in medicine. Its water insolubility and high metabolism rate require the use of drug delivery systems to make it effective in the human body. Among various types of nanocarriers, block copolymer based ones are the most effective. These polymers are broadly used as drug-delivery systems, but the nature of this process is poorly understood. In this paper, we propose a molecular dynamics simulation study of the interaction of Curcumin with block copolymer based on polyethylene oxide (PEO) and polypropylene oxide (PPO). The study has been conducted considering the smallest PEO and PPO oligomers and multiple chains of the block copolymer Pluronic P85. Our study shows that the more hydrophobic 1,2-dimethoxypropane (DMP) molecules and PPO block preferentially coat the Curcumin molecule. In the case of the Pluronic P85, simulation shows formation of a drug–polymer aggregate within 50 ns. This process leaves exposed the PEO part of the polymers, resulting in better solvation and stability of the drug in water.
Curcumin, a naturally occurring drug molecule, has been extensively investigated for its various potential usages in medicine. Its water insolubility and high metabolism rate require the use of drug delivery systems to make it effective in the human body. Among various types of nanocarriers, block copolymer based ones are the most effective. These polymers are broadly used as drug-delivery systems, but the nature of this process is poorly understood. In this paper, we propose a molecular dynamics simulation study of the interaction of Curcumin with block copolymer based on polyethylene oxide (PEO) and polypropylene oxide (PPO). The study has been conducted considering the smallest PEO and PPO oligomers and multiple chains of the block copolymer Pluronic P85. Our study shows that the more hydrophobic 1,2-dimethoxypropane (DMP) molecules and PPO block preferentially coat the Curcumin molecule. In the case of the Pluronic P85, simulation shows formation of a drug–polymer aggregate within 50 ns. This process leaves exposed the PEO part of the polymers, resulting in better solvation and stability of the drug in water.
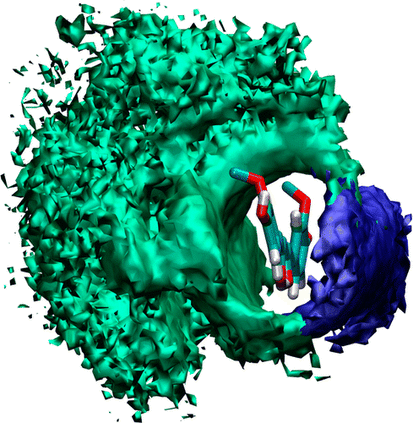
Fig: Spatial distribution of PEO and PPO chains around the Curcumin molecule. PEO is shown in blue and PPO in red. The density surfaces have an isovalue of 250 for both PEO and PPO.
